
二元层状过渡金属硫族化合物通常可以由分子式MX2表示,其中,M为正四价的过渡金属(IVB-VIIB),X为负二价的硫族化合物S、Se和Te。由于原子排布的不同, MX2块体性质各异,可以表现出绝缘体、半导体、半金属或金属。当把这些块材剥离成单层或者少层时,由于纳米局限效应的存在,在保存原有性质的同时,会出现新的物理和化学行为。
与此同时,碳元素位于元素周期表IVA,其单质有多种同素异形体,例如零维的富勒烯(C60)、一维的碳纳米管(CNTs)、二维的石墨烯(Graphene)和三维的金刚石等。其中,CNT和Graphene由于其独特的表面和微结构,在能源领域方面具有较好的应用潜能。
近期,中国科学技术大学国家同步辐射实验室宋礼教授课题组利用MX2层状结构和碳纳米材料的优势,合成出具有竖直排列金属相的MoX2(X=S/Se)复合纳米结构,实现了离子电池性能的大幅提升。相关成果发表于国际著名期刊ACS Nano (2017, 11, 6483-6491)和Nanoscale (2017, 9, 6975-6983),文章第一作者是硕士研究生项婷。
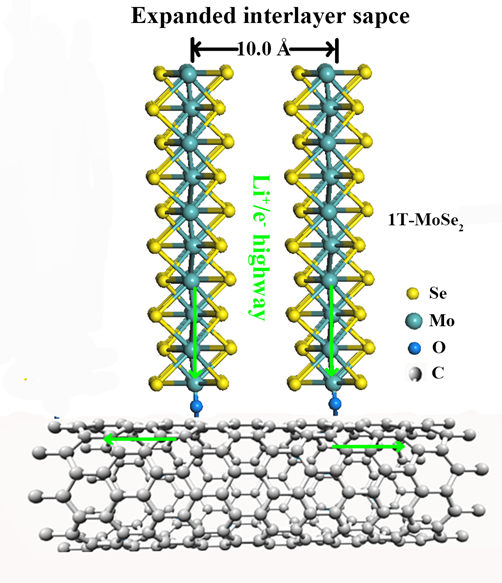
示意图:竖直1T-MoSe2复合SWCNTs结构在充放电过程中的电化学过程
该研究采用溶剂热法成功合成出具有扩大层间距的金属相1T-MoSe2纳米片竖直生长在SWCNTs表面具有独特结构的复合物。同步辐射XAFS和XANES表征结果表明,SWCNTs表面上的的含氧基团为1T-MoSe2纳米片的生长提供活性位点,并以C-O-Mo键形式与1T-MoSe2纳米片边缘的Mo原子键合。1T-MoSe2和SWCNTs之间的强C-O-Mo键结合,确保了结构的稳定性,防止充放电过程中的结构垮落。此外,1T-MoSe2/SWCNTs复合物薄膜具有优异的锂离子电池性能,在300mA/g的电流密度下循环100圈后比容量高达971mAh/g。高导电性SWCNTs和金属性的1T-MoSe2纳米片构成了高效的电子/离子传输通道,即使在3000mA/g的电流密度下,1T-MoSe2/SWCNTs复合物依然具有630mAh/g的比容量。详细实验结果请参考ACS Nano (2017, 11, 6384-6491)。
随后,这种原位合成方法被应用于构筑其他二维复合结构,实现了金属相MoS2纳米片在氧化石墨烯表面的竖直生长。研究结果表明,合成出的1T-MoS2/graphene复合物在充放电过程中能够有效抑制锂离子嵌入/脱出造成的体积变化,防止结构的损毁,维持结构的完整性。此外,高导电的石墨烯与竖直排列的金属相硫化钼共同组成导电网络为电子和离子提供高速传输通道,从而获得更加优异的倍率性能,在高电流密度3500mA/g下,依然能够展现出666mAh/g的比容量。详细实验结果请参考Nanoscale (2017, 9, 6975-6983)。
这些研究结果不仅为多种维度纳米结构的理性设计和原位合成提供了一种新的思路,也揭示了纳米复合结构中不同组分之间的协同效应,为理解纳米结构和性能之间的内在联系提供了新的方法。
以上研究工作得到了科技部重点研发计划和青年973计划、国家自然科学基金、合肥大科学中心、纳米科学卓越中心、南开大学先进能源材料化学教育部重点实验室等项目和机构的资助。

