
分子模拟是物质微观结构演化、光谱学模拟、材料构效关系、化学反应机理等科学研究的重要理论模拟工具。分子模拟的核心问题是根据原子几何坐标计算体系的势能和势能梯度,传统上这类计算一般依赖经验势方法或者第一性原理方法(如密度泛函方法DFT等)。一般来说,经验势方法计算效率高,可用于大体系长时间过程的模拟,但模拟精度较低,且难以描述化学键的形成和断裂等反应过程;第一性原理方法具有较高的物理精度,可描述化学反应,但计算成本很高,模拟的空间尺度一般局限在两三百原子以内,时间尺度局限在皮秒量级。因此,发展具有第一性原理级别模拟精度和经验势级别计算资源需求的分子模拟方法,对大尺寸体系和长时间尺度的过程模拟(如相变、化学反应等)至关重要。中国科学技术大学国家同步辐射实验室XMCD线站团队的王超副研究员和中国科学技术大学信息科学技术学院的冯亚娟特任副研究员合作,发展了一种基于机器学习的分子模拟方法,模拟精度接近DFT计算,计算耗时降低四个数量级以上且具有线性标度;利用该方法研究了经典体系三氧化硫复杂水解反应微观机制。相关研究成果以“Surface Confinement of Finite-Size Water Droplets for SO3 Hydrolysis Reaction Revealed by Molecular Dynamics Simulations Based on a Machine Learning Force Field”为题于5月2日发表在《美国化学会志》杂志(J. Am. Chem. Soc. 2023, 145, 10631, DOI: 10.1021/jacs.3c00698)。
王超副研究员和冯亚娟特任副研究员从2020年开始合作开发机器学习驱动的分子模拟方法,基于对称函数局域结构特征提取和人工神经网络的高维势能面描述方法,开发了具有自主知识产权的机器学习分子模拟程序(Nanoscale 2021, 13, 12212; 软件著作:2021SR0185084)。最近,研究团队将机器学习模型进一步扩展到应用到化学反应过程的高精度模拟,利用对称函数提取体系中每个原子的局域结构特征,训练人工神经网络实现从局域原子结构向“原子势能”的映射。这种设计使模型天然具有“反应性”,可描述化学键的形成和断裂过程。在软件架构上,基于GPU平台的异构计算进一步提高了数值计算效率。为了提高模型训练效率,研究团队发展了融合了Metadynamics、伞形采样、分子动力学采样和蒙卡采样的机器学习模型训练数据集构建方法,实现了复杂化学反应空间中的高效精准采样。以经典的三氧化硫水解反应体系为例,利用机器学习模型对三氧化硫分子和不同尺寸水团簇的水解反应体系进行了大量反应轨迹采样,系统揭示了三氧化硫分子水解的不同反应分支(分别生成中性(H2SO4)−(H2O)团簇、(HSO4)-−(H3O)+离子团簇、(SO4)2-−[(H3O)+]2离子团簇)来源于有限尺寸水团簇的表面限域效应。
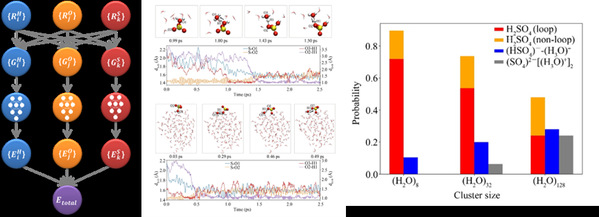
左:机器学习分子模拟模型原理结构;中:三氧化硫-水体系的部分反应轨迹;右:大量反应轨迹总结的反应分支比
本项研究展示了机器学习驱动分子模拟方法应用于复杂反应体系模拟的潜力,在后续研究中,将基于该模型建立复杂动态体系谱学模拟方法,实现原位谱学实验的“数字孪生”,应用于未来海量同步辐射谱学数据的高通量分析。
王超副研究员和冯亚娟特任副研究员为论文共同通讯作者。该项研究工作得到了国家自然科学基金,合肥大科学中心高端用户培育基金和中国科学技术大学重要方向项目培育基金等项目经费资助。

