含氮有机化合物,特别是那些含有羰基胺部分的化合物,在药物合成、化学品和先进聚合物制造中是关键的中间体。这一现状激发了电催化C-N偶联策略的发展。目前的研究主要集中在CO2、CO与无机氮源(如氨、硝酸盐或亚硝酸盐)的偶联以生产单碳产品,如尿素、甲胺和甲酰胺。值得注意的是,乙酰胺衍生物(CH3−C(=O)−NR'R')作为有机试剂、农用化学品和染料的通用基石,有着广泛的应用和巨大的需求,但它们的工业规模生产仍然依赖于在高温、高压酸性条件下使用乙酸酐/酸的高能耗热化学路线。通过电化学CO还原进行胺的电催化乙酰化是一种引人注目的绿色替代方法,但也面临着内在的科学障碍:复杂的多电子转移过程和相互竞争的反应途径通常会导致乙酰胺衍生物的性能不佳和选择性有限,这给催化剂设计和反应微环境控制带来了根本性的挑战。
通过CO进行的胺类电催化乙酰化反应一般有两步机制:(1)CO的电化学C-C偶联生成C2中间体,(2)胺类在活性碳上亲核攻击,最终形成C-N键。虽然之前的研究已经建立了Cu+稳定策略(通过金属掺杂或表面改性)作为C-C偶联的有效促进剂,但随后的C-N偶联(主要发生在电极-电解质纳米限制界面)需要精确的界面微环境控制。例如,水合阳离子的大小是影响界面的关键参数,阳离子的溶剂化效应不仅可以调节界面局部pH和CO2/CO浓度,还可以促进C-N偶联过程。相反,与阳离子相比,卤化物阴离子表现出独特的界面行为:它们在铜催化剂上的强化学吸附诱导了双重功能——同时改变了局部质子活性(通过亥姆霍兹层重构),并通过表面重构重塑了催化剂形态。然而,阴离子介导的电子相互作用的原子机制及其在加速C-N偶联动力学中的具体作用仍然没有得到充分的了解。
湖南大学王双印教授/邹雨芹教授团队依托合肥光源红外谱学与显微成像线站发展的原位显微电化学红外光谱技术,在电催化合成二甲基乙酰胺领域取得新进展。我们开发了一种卤化物介导的微环境调节策略,通过在氧化亚铜底物上碘化物协同化学吸附,建立了一个动态的Cu0/Cu+界面,有效抑制Cu2O的还原,同时促进CO和二甲胺之间高效的电催化C-N偶联,用于二甲基乙酰胺的合成。优化后的电解质和催化剂具有良好的性能,DMAC的法拉第效率为45.6%,最大产率为435.9 mmol·gcat.-1·h-1,选择性70%。值得注意的是,该策略证明了合成各种乙酰胺衍生物的通用性。这一突破为利用CO2/CO作为可持续碳原料的电化学合成增值酰胺建立了范例,为碳排放增值提供了切实可行的解决方案。
通过原位同步辐射傅里叶变换红外光谱(图1),在1000~1200 cm-1的光谱范围内出现了C-OH拉伸振动,表明*COCOH中间体的初始形成,表明碘化物显著促进了C-C偶联过程。随后,光谱显示在约1613 cm-1处对应C=O键的强化吸收峰。另外,在1314 cm-1处出现一个特征性的*CH3峰,进一步证明了乙酰基的形成。在1401 cm-1处有一个突出的峰,这是由于C-N键的形成,在碘化物的存在下表现出增强的强度,突出了它在促进C-N偶联反应中的重要作用,而C-N偶联反应是合成DMAC的重要前提。基于这些观察,可以得出结论,碘离子具有双重作用:它不仅促进催化剂表面C-C偶联生成C2中间体,而且促进含碳中间体与DMA的偶联,从而实现关键的C-N键的形成。
此外,还进行了DFT计算(图2),以确定在Cu和Cu/Cu2O-I-表面整个反应路径上的能垒。CO分子最初吸附在催化剂表面。随后,两个CO分子通过电化学C-C键合过程结合形成*COCOH中间体,这是生成C2产物的关键步骤,也是该反应的速率决定步骤。碘离子的存在显著降低了这种偶联反应的能垒,从0.67 eV降低至0.35 eV,这与红外光谱结果相符。通过*COHCOH的电化学脱水生成了酮烯中间体*CCO,随后由HNMe2进行亲核攻击生成*CCONHMe2中间体。在C-N偶联之后,*CCONHMe2发生质子迁移形成*CHCONMe2。最后,通过两步电化学氢化和随后的解吸生成了DMAC。这些发现表明,碘离子的掺入不仅促进了CO的初始C-C偶联,而且还降低了*CCO和HNMe2偶联的反应势垒。
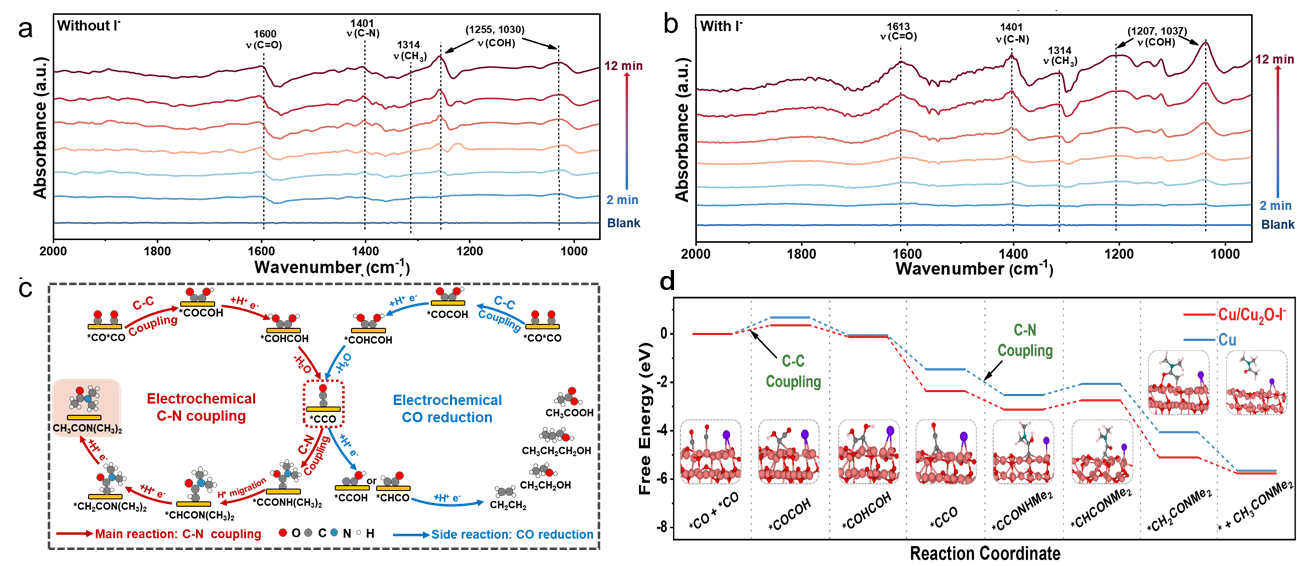
图1. 原位同步辐射傅里叶变换红外光谱结合DFT计算确认CO电化学还原偶联二甲胺合成二甲基乙酰胺的反应路径
相关研究成果以“Electrochemical coupling of carbon monoxide and amine on iodide coordination stabilized Cuδ+ site.”发表在国际著名学术期刊《Nature Communications》上。论文共同第一作者为樊赟, 鄢云辉, 安琦正。


